

 Picture from his faculty page - © F.Stuhl |
Werner Kutzelnigg was born in 1933. He studied chemistry in Bonn,
Freiburg (Germany). He then was postdoc with Pullman and Berthier
1960-1963 and with Lowdin in Uppsala. Since 1970 professor in Karlsruhe, then from 1973-1998 in Bochum, Germany. Prof. Emeritus. He enters in this context since he has written a 2 volume 'Introduction to Theoretical Chemistry' book which in Germany and as well as in many other countries (translations) has been very widely in use and which in 1994 appeared in a second edition. |
Ruhr-Universität Bochum, 24.07.1997; 09:15
Anders: Herr Professor Kutzelnigg, wenn ich einleitend aus der Literatur (1) ein paar Daten zu Ihrem Werdegang erwähnen darf:
Geboren 1933 in Wien. Sie studierten Chemie in Bonn und Freiburg, promovierten 1960 bei R. Mecke über ein Thema aus der IR-Spektroskopie (2). 1960-1963 waren Sie Postdoc bei B. Pullman und G. Berthier in Paris und in Uppsala bei P.O. Löwdin. Habilitiert 1967 in Göttingen, 1970 Professor in Karlsruhe, ab 1973 in Bochum. Sie haben ein bekanntes zweibändiges Lehrbuch geschrieben, 1975 und 1978, das 1992 bzw. 1994 in zweiter Auflage erschien (3).
Was hat Sie damals bewogen, nach Paris zu gehen?
Prof. Kutzelnigg: Ich hatte in Freiburg, als ich noch an meiner Doktorarbeit war, einen Vortrag von Bernard Pullman gehört, der mich sehr beeindruckt hat, da habe ich zum ersten Mal die Botschaft erhalten, daß man mit quantenchemischen Methoden etwas praktisch machen kann. Ich wußte noch relativ wenig darüber und hatte zwar gehört, daß man die chemische Bindung verstehen kann, wenn man die Quantenmechanik einsetzt. Daß man aber wirklich praktische Anwendungen machen kann - - bei ihm ging es vor allem um pharmazeutische Anwendungen, Antimetabolismus und dergleichen - - das hatte mich beeindruckt und da habe ich beschlossen, zu Pullman zu gehen. Später habe ich allerdings gemerkt, daß der Optimismus, den Pullman ausgestrahlt hat, doch wohl etwas verfrüht war. Aber ich habe es insgesamt nicht bereut, nach Paris gegangen zu sein.
A: Sie haben in Paris sofort theoretisch gearbeitet (4-6). Eigentlich mußten Sie als physikalischer Chemiker ja erst einmal den gesamten mathematischen Apparat der Quantenmechanik lernen?
K: Also da war ich schon ganz gut vorbereitet. Ich hatte bereits während meines Studiums Vorlesungen in Theoretischer Physik gehört, ich hatte den ganzen theoretischen Zyklus absolviert. Dann war ich auch in der Sommerschule für Quantenchemie in Uppsala - -
A: Aha, ja, vorher schon - -
K: Da war ich vorher schon. Insofern war ich da ganz gut vorbereitet.
A: Wie sind Sie an das Geld herangekommen? Das waren alles NATO-Stipendien?
K: Naja, das ist richtig. Für die Teilnahme an dieser Sommerschule in Uppsala habe ich ein OECD-Stipendium gehabt und dann in Paris ein NATO-Stipendium. Nun, ich glaube, heute kann man wieder ohne Probleme erwähnen, daß man das hatte. Ich erinnere mich, als ich zu einem Vorstellungsvortrag in Erlangen war, daß ich da von einem Studentenvertreter angesprochen wurde, auf das NATO-Stipendium - ob ich da nicht sozusagen Kriegsforschung betrieben habe. Das war natürlich überhaupt nicht der Fall. Es gab selten Stipendien, die mit so wenig Auflagen verbunden waren, wo man so frei in der Forschung war wie bei diesen NATO-Stipendien. Es war eine tolle Sache und solche Vorstellungen, wie sie Studenten in der '68-er Zeit damals möglicherweise damit verbunden haben, waren absolut abwegig.
A: Arbeitete man bei den Pullmans Tag und Nacht? Es gab doch so viele Veröffentlichungen. Gerade die Pullmans waren doch sehr aktiv.
K: Ja wissen Sie, damals gab es diese elektronischen Rechenmaschinen noch nicht, wie man sie heute hat und Herr Pullman hatte einen technischen Mitarbeiter, einen Monsieur Astic, der auf einer Friden, einem mechanischen Rechner, die ganzen Hückel-Rechnungen gemacht hat. Und zwar hatte diese spezielle Maschine eine automatische Quadratwurzel. Astic hat von morgens bis abends mit der Maschine gerechnet. Er hatte sich besondere Tricks ausgedacht, die Säkulargleichungen zu lösen, die Nullstellen von den Hückel Polynomen zu finden. Und zum Normieren der Eigenvektoren hatte er also eine Quadratwurzel.
A: Rechnete er immer fehlerlos?
K: Ich glaube schon, er war sehr zuverlässig,. Er hat die Ergebnisse vermutlich auch auf andere Weise abgecheckt. Da gibt es ja eine ganze Reihe von Tests: die Summe der Eigenwerte einer Matrix muß gleich der Summe der Diagonalelemente (der Spur), in diesem Falle Null, sein. Die haben auch Moleküle mit Heteroatomen gerechnet, da war dann dir Spur nicht immer gleich Null. Und ich habe irgendwo noch diese dicken Bände von Pullman, wo sie alles, was es so gab, nach Hückel durchgerechnet haben. Der arme Astic, als die elektronischen Rechner kamen, da war er ziemlich beschäftigungslos, etwas anderes konnte er eigentlich nicht, und er hat dann mehr oder weniger Daumen drehen müssen.
A: Die IBM 1620 war relativ klein (7), Sie haben mit SPS (Standard Programming System) gearbeitet. War das eine Art Makrosprache?

K: Also, das ist so. Die IBM 1620 war eine ganz eigenartige Maschine. Sie war eine der ersten, sie kam relativ bald nach der 650. Die 650 war eher ein Prototyp, auf der dann die anderen aufgebaut haben, es war auch eine Maschine, die mit Wörtern gearbeitet hat. Dagegen war die 1620 eine Maschine, wo das Element nicht ein Wort war, sondern eine Dezimalziffer. Man hat mit Dezimalziffern gearbeitet, hat nicht binär gerechnet, und Multiplikation und Division wurden durch Tabellen für Dezimalziffern ausgewertet. Es war nur fixed point vorhanden, dazu gab es eine Software, das war vermutlich das SPS, damit konnte man dann mit floating points auf Softwarebasis arbeiten, mit beliebiger Stellenzahl allerdings. Sie konnten also selber entscheiden, welche Genauigkeit Sie wollten, mußten das entsprechend festlegen, was ich dann für gewisse Rechnungen ausgenützt habe, erforderlich war eine hohe Genauigkeit. Aber es gab keine peripheren Speicher, den gab es überhaupt nicht. Es gab einen internen Kernspeicher, der war sehr klein - -
A: 20 KB.

K: Ja. - und für alles Periphere mußte man Lochstreifen benutzen. Ich habe also die Zwischengrößen auf Lochstreifen gespeichert und später wieder eingelesen. - Es war ganz lustig. Aber immerhin waren da gewisse Sachen möglich.
A: Einen card punch hatten Sie nicht?
K: Nein, nur Lochstreifen.
A: Laut IBM gelocht mit 15 Zeichen pro Sekunde, gelesen mit 150 Zeichen pro Sekunde. Und Sie haben mit dieser Maschine quantenchemische Rechnungen gemacht (5)?
K: Ich habe unter anderem Rechnungen am Heliumatom gemacht (6). Es war, - ja - , ganz lustig.
A: Haben Sie diese Sachen auf der IBM 1620 gerechnet?
K: Ja, die Rechnungen für die ersten papers.
A: Und 1966, was mich in meinem Zusammenhang eben interessiert, hatten Sie dieses Hückel Problem der alternierenden/nicht alternierenden Bindungen bearbeitet (8) - -
K: Da hab ich auch mal was drüber gemacht - ja.
A: War das - wie soll ich sagen - eine Gefälligkeitsarbeit für Pullman?
K: Das war schon in Göttingen, als ich das gemacht habe.
Das Thema war ja aktuell. Sagen wir mal, da haben sich Leute wie Heilbronner damit beschäftigt, und andere. Es ist so: Eigentlich war die Arbeit, die ich gemacht habe, nicht so grundsätzlich neu. Denn das Problem der Beschreibung der Bindungsalternierung bei Polyenen, Ringpolyenen, ist ja schon lange vorher von Coulson einerseits und Lennard-Jones andererseits gelöst worden. Aber ich wollte zeigen, daß man die Trends, die man experimentell beobachtet, mit der Störungstheorie ganz gut hinbekam.
Meine Idee war, daß man bei alternierenden Systemen zusätzlich zu der topologischen Matrix, die die Nachbarschaftsverhältnisse definiert, noch eine Alternierungsmatrix einführt. Da braucht man einen Alternierungsparameter und dann liegt es nahe, den Effekt der Alternierung störungstheoretisch zu berechnen.
Ich habe das in meinem Buch noch ein bißchen ausgedehnt (29). Ich glaube nicht, daß es eine große praktische Bedeutung hat, aber es ist sozusagen ein wichtiger Schritt über die einfache Hückel -Näherung hinaus. Der wichtigste Fehler der Hückel-Näherung liegt bei Polyenen darin, daß die Alternierung vernachlässigt wird, die aber sehr wichtig ist. Das war also mein Interesse. Ich habe mich immer schon für einfache formale Dinge interessiert - - aber das war sozusagen nicht mein Hauptarbeitsgebiet.
A: Darf ich noch eine Frage zu Paris stellen: Wie hat es Bernard Pullman gefallen, daß Sie da jetzt theoretisch gearbeitet hatten?
K: Ach - es hat ihm nicht sehr gefallen, würde ich sagen. Aber da ich mein eigenes Geld mitgebracht hatte, wie es so schön heißt, konnte er mir ja keine Vorschriften machen. Ich hatte ursprünglich durchaus die Absicht gehabt, auf dem Pullman'schen Gebiet zu arbeiten. Ich hatte Ihnen das schon gesagt. Was mich zunächst beeindruckt hat, war die Arbeit von Mme. Pullman und Nakashima, um Antimetaboliten zu entwickeln, Purinbasen, analoge oder ähnliche Dinge. Aber dann habe ich gemerkt, daß es mich doch nicht so überzeugt hat und ich bin - auch vielleicht unter dem Einfluß der Sommerschule in Uppsala - dann doch sehr stark in die ab initio Theorie geschwenkt. Das lag nicht auf der Pullman-Linie. Aber wie gesagt, ich hatte meine eigenes Geld und - -
A: Gab es Seminare bei den Pullmans? Waren die Pullmans in die Lehre eingebunden?
K: Sie haben, soviel ich weiß, keine Vorlesungen an der Uni gehalten. In die Lehre eingebunden war z.B. Mme. Serre, die hat in der ENS des Jeunes Filles auch Vorlesungen über theoretische Chemie gehalten. Daudel hatte auch wenig mit der Lehre zu tun. Also die Lehre in Theoretischer Chemie spielte in Paris eine geringere Rolle als die Forschung.
A: Das erklärt natürlich die Vielzahl von Veröffentlichungen, die da im Gebiet der Quantenchemie in dieser Zeit aus Frankreich kamen.
K: Ich würde mir keine Gedanken machen zur Erklärung der Zahl der Veröffentlichungen. Diese hat so viele Gründe. Ein bißchen rührt sie daher, daß einfach die Zahl von Veröffentlichungen überbewertet wird. Einmal sind die Leute bestrebt, möglichst viele Publikationen zu schinden. Für mich war das nie ein wesentliches Qualitätskriterium gewesen. Ich habe noch nie einen Wissenschaftler nach der Zahl seiner Publikationen gefragt. Mich kann man damit nicht beeindrucken. Aber es gibt Leute, die das bewegt.
A: Daudel hat von 50 Mitarbeitern erzählt (9). So entstehen natürlich viele Veröffentlichungen. Und dann gibt es die slave drivers - -
K: Es gibt Leute, die den Ruf eines 'slave drivers' haben. Aber Daudel war kein 'slave driver'. Daudel war immer schon etwas abgehoben. Das Daudel'sche Institut galt als recht attraktiv. Vielleicht weil es aus der Sicht der Quantenchemie vielseitiger war als das, was Pullman machte, der war doch auf biologische Anwendungen festgelegt. Bei Daudel sind immer eine ganze Reihe von verschiedenen Themen aus der Theoretischen Chemie bearbeitet worden. Und sein Institut hat immer sehr viele Gäste angezogen, vor allem Amerikaner. Ich würde sagen, die sind in erster Linie dahin gegangen, weil sie ganz gerne mal nach Paris wollten. Es war eine sehr gute, eine sehr internationale Atmosphäre im Daudel'schen Institut. Und deshalb war ich dort oft Gast, ich bin zu den meisten Seminaren gegangen. Ich hatte eine ganze Reihe von Leuten kennengelernt, die ich in der Zeit, wäre ich in Amerika gewesen, nie hätte kennenlernen können. Ich könnte eine ganze Menge Namen nennen von Amerikanern, die ich kennengelernt habe. Übrigens gab es auch im Pullman'schen Institut interessante Gäste, z.B. John Platt.
A: Kleine Nebenfrage: brachten die alle ihr eigenes Geld mit?
K: Ich vermute ja. Daudel hatte wohl auch Geld, um Leute einzuladen. Es waren eine ganze Menge von Amerikanern, die Rang und Namen hatten, die man da immer in Paris antreffen konnte.
A: Eine ganz andere Frage: Herr Prof. Kutzelnigg, was ist eigentlich Semiempirie?
K: Semiempirie heißt eigentlich: zur Hälfte Empirie. Gemeint ist, daß man von einem Ansatz, ursprünglich von einem quantenchemischen Ansatz ausgeht, der im Prinzip richtig ist, aber doch sehr genähert, und man kompensiert die Fehler dieser Näherung dadurch, daß man gewisse Größen nicht wirklich ausrechnet, sondern als wählbare Parameter ansieht, die man dann an das Experiment anpaßt. Im Falle der Hückel Theorie ist das eigentlich besonders schön. Da hat man bei Kohlenwasserstoffen gerade zwei Parameter, a und b , und die wählt man halt so, daß eine Reihe von experimentellen Größen richtig beschrieben werden - und dann kann man ganz gut interpolieren. Dabei ist es ganz lustig, gerade bei diesen b -Parametern, daß man unterschiedliche b 's nehmen muß - für thermochemische Größen und für spektroskopische Größen. Also etwa 1 eV für themochemische Größen und 4 eV für spektroskopische Größen. Also das zeigt schon, ein bißchen, daß etwas unbalanciert ist. Aber das ist sozusagen die Bedeutung von semiempirisch verglichen mit ab initio.
Es gibt dann noch die rein empirischen Methoden, das sind z.B. die Kraftfeldmethoden, wo man von vornherin nichts aus ab initio Theorien nimmt, sondern wo man irgendwelche Ansätze macht für Kraftfelder, harmonisches Kraftfeld, mit Kopplungskonstanten und ähnlichem. Das paßt man alles an das Experiment an. Das sind die rein empirischen Methoden.
Ein boshafter englischer Kollege - ich glaube Guggenheim - hat mal den Begriff sesquiempirisch definiert, also in dem Sinne: bei der semiempirischen Methode hat man halb soviel Parameter wie Größen, die man beschreiben will. Bei den rein empirischen Methoden hat man soviel Parameter wie man Größen beschrieben will. Bei den sesquiempirischen Methoden hat man 1.5 mal soviel Parameter wie zu berechnende Größen.
A: Coulson soll mal gesagt haben: vor dem Essen glaube er an gewisse Parameter, nach dem Essen wieder weniger (10).
K: Naja, Coulson war ja auch Engländer, die neigen oft so ein bißchen zu humoristischen Formulierungen. Ich meine, man sieht die Sache heute ja anders, das muß man ganz deutlich sagen. Man hat früher die Hückel Methode wirklich als Näherungsmethode zur Lösung der Schrödingergleichung angesehen. Das würde man heute nicht mehr tun, man würde sagen: das ist eben ein Modell, das gewisse Trends der Realität richtig wiedergibt - und so soll man es sehen und das hat natürlicherweise seine Grenzen. Es hat aber natürlich auch eine ganze Reihe Vorteile: eben den der formalen Einfachkeit. Und man kann auch leichter sehen, wo es dann nicht funktioniert. Das muß man ja auch ganz ehrlich sagen, dann lernt man etwas wichtiges, gewissermaßen, wenn man sieht, wo ein Modell versagt.
A: Deswegen haben Sie das ja in Ihrem Buch (3 a-b) relativ stark herausgestellt. Ich suche eben die Chemie, die Modelle dazu, in der Quantenchemie. Einer der Arbeitstitel ist: wie kam das Quant in die Chemie. Das andere Problem, das ich als Chemiker sehe ist, daß die Quantenchemie unsere Chemie hernehmen will, um damit nur zu rechnen. Und letzlich lassen die Theoretiker den praktischen Chemiker dann im Stich. Die Quantenchemie abstrahiert so weit, daß der Chemiker dann nicht mehr folgen möchte.
K: Das will er ja auch nicht. Wie sich die numerische Quantenchemie heute entwickelt hat, liefert sie Zahlen. Sie liefert Ergänzungen zum Experiment. Die Interpretation ist heute nicht mehr die Hauptaufgabe der Quantenchemie. Man möchte vor allem Vorhersagen machen, man möchte etwas rechnen, weil es billiger ist als ein Experiment zu machen und das funktioniert oft wunderbar. Das ist eine ganz andere Entwicklung, als man früher vielleicht einmal gedacht hat.
A: Als Chemiker suche ich gelegentlich Auskunft bezüglich Maxima bei Spektren in einem Programm namens HyperChem (11), um z.B. in der Flüssigchromatographie einer bestimmten Verbindungsklasse die Geräteparameter einzustellen. Die erhaltene Auskunft ist nicht immer korrekt.
K: Nun, HyperChem ist natürlich ein Programm, das im wesentlichen sozusagen auf Korrelationen von Experimenten beruht, etwa auf der Basis von neuronalen Netzen oder dergleichen Dingen, nicht wahr. Es ist keine ab initio Theorie.
A: Es beinhaltet eine Reihe von semiempirische Methoden.
K: Ja, und sucht eben dann nach Korrelationen, so etwa im Sinne, so wie man es auch mit neuronalen Netzen macht: man sucht nach irgendwelchen nichtlinearen Korrelationen, man geht von einem Trainingsset aus, man wendet es auf ein Testset an und wenn das irgendwann gut funktioniert, dann läßt man es auf eine Verbindungsklasse los. Aber diese Methode ist außerordentlich billig im Rechenaufwand. Bei ab initio Methoden ist der Rechenaufwand viel größer, dementsprechend ist die Aussagekraft in der Regel auch größer. Sie ist natürlich auch durch die Größe der Moleküle eingeschränkt.
Zu Ihrer Frage, wie das Quant in die Chemie kommt: Die Frühgeschichte der Quantenchemie war ja auch etwas eigenartig. Sie ist dominiert worden durch Pauling. Und Pauling war sehr stark von der Chemie, von der chemischen Intuition, geprägt. Er hat zwar auch eine Menge von Quantenmechanik verstanden, aber er war doch eher ein Mann, der von der Chemie her kommt (12). Und er hat versucht, für die Vorstellungen, die man in der Chemie damals schon entwickelt hatte, die Mesomerielehre und ähnliche Dinge, ein quantenmechanisches Pendant zu finden. Es war also nicht so, daß er von der Theorie ausging und versuchte, sozusagen deduktiv auf die Chemie zu kommen. Das war damals undenkbar, heute macht man das. Meiner Ansicht nach ist die Zeit von Pauling lange vorbei. Er hat dominiert und teilweise auch vielleicht die Entwicklung behindert.
A: Wo könnte der Begriff Pauling Point (13) herkommen?
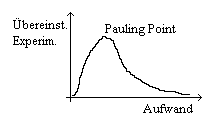
K: Ich nehme an von Löwdin. Er hatte folgendes Diagramm gemacht: Da hat er aufgetragen, ich glaube, Übereinstimmung mit dem Experiment als Ordinate und als Abszisse Aufwand. Und da hatte man sozusagen zunächst mal einen Peak für wenig Aufwand, eine sehr hohe Übereinstimmung mit dem Experiment, und dann dümpelt das dann irgendwo weiter unten. Diese gute Übereinstimmung, mehr oder weniger zufällig, mit wenig Aufwand, das war der Pauling Point. Also ich nehme an, daß der Begriff auf Löwdin zurückgeht. Ich glaube nicht, daß dem Pauling der Begriff Pauling Point sehr sympathisch war.
|
Periode |
Zeitraum |
Beschreibung |
Namen, z.B. |
|
1 |
1927-1935 |
Grundlegendes |
Hund, Hückel, Hellmann |
|
2 |
1935-1950 |
Übergang |
Mulliken, Coulson |
|
3 |
1950 - jetzt |
Computer |
Roothaan, Boys, Pople |
A: Sie haben in Ihrem Buch (14) folgende Zeitgliederung für die Entwicklung der Quantenchemie angegeben. Würden Sie, im großen und ganzen diese Perioden, geschichtlich gesprochen, immer noch so bebehalten?
K: Ich denke eigentlich schon. Die 3. Phase geht jetzt so kontinuierlich weiter. Natürlich gibt es gewisse Änderungen der Trends, aber die Numerik dominiert. Sagen wir mal, die Interpretation, die qualitative Interpretation, ist sehr stark in den Hintergrund getreten. Das machen nur relativ wenig Leute (unter denen insbesonders Klaus Ruedenberg zu nennen ist) und man bemüht sich eben wirklich, komplementär zum Experiment zu sein, eben quantitativ.
A: Ein Komplement zum Experiment oder eine reale Umgehung des Experiments, das zu teuer ist?
K: Die Computerchemie sieht sich durchaus auch als Konkurrenz zum Experiment. Es wird sich herausstellen, im Laufe der Zeit, daß Rechnungen relativ zum Experiment immer billiger werden. Und wenn Sie die Eigenschaften eines Moleküs vorhersagen können, und feststellen, das sind nicht die, die Sie wollen, dann brauchen Sie es erst gar nicht zu synthetisieren. Das ist sicher ein Trend, der zu erkennen ist.
Man spricht heute vielfach von Computerchemie. Computerchemie bedeutet eigentlich, daß man die Methoden als vorhanden ansieht und daß die Anwendungen im Vordergrund stehen. Die Methoden liegen vor als black boxes, die jeder benutzen kann. Aber parallel dazu geht natürlich die Methodenentwicklung weiter, das darf man dabei nicht vergessen. Und man muß sich, je nachdem worauf man hinaus will, neue Dinge ausdenken. Auf dem Gebiet der Genauigkeit, das ja auch eine Rolle spielt, im Zusammenhang mit Spektroskopie, wird es dann zunehmend wichtig, gewisse, bisher vernachlässigte Effekte zu berücksichtigen: beyond-Born-Oppenheimer, Relativistik und dergleichen.
Es gibt in einem ganz anderen Bereich Leute, die sich bemühen, die Anwendbarkeit der Theorie auf sehr große Moleküle zu erweitern. Das ist wichtig für Proteine, andere Biomoleküle, Stichwort Molecular Modeling, ist aber natürlich von dem, was die ab initio Quantenchemie macht, relativ weit entfernt.
A: Und dann die Frage der Solvation. Ist das auch etwas, was man ab initio machen kann?
K: Ja, natürlich. Es geht um zwischenmolekulare Wechselwirkungen, Solvationseffekte sind auch so etwas. Eine ganz große Rolle spielen Untersuchungen an Oberflächen. Da hat man sozusagen Berührungen mit der Festkörperphysik. Der Festkörper ist eigentlich immer noch eine Domäne der Physik, aber auch die Chemie dringt immer mehr in das Gebiet der Festkörper ein, aus verschiedenen Gründen. Teilweise vom Cluster herkommend: Cluster sind also ganz kleine Festkörper, das machen die Chemiker. Und wie gesagt, Oberflächen. Lösungen werden sicher auch zunehmend auch eine Rolle spielen.
A: Ihr Artikel in Fortschritte der Chemischen Forschung von 1970 mit Del Re und Berthier (15). Da hatten sie das s /p -Problem untersucht. Wie war übrigens die Reaktion der organischen Chemiker auf diesen Artikel? Hatten Sie da ein Feedback?
K: Schwer zu sagen. Es war die Zeit, wo man doch noch sehr stark in der Verteidigungsposition war. Vielen organischen Chemikern mußte man erst einmal klar machen, daß Theoretische Chemie überhaupt irgend einen Sinn hat. In diesem Artikel ging es die s - und p -Elektronen. Das war ein Thema, das etwas kontrovers diskutiert wurde. Uns ging es darum zu klären, was es eigentlich bedeutet, wenn man von s - und p -Trennung spricht. Das haben verschiedene Leute verschieden gesehen. Preuss z.B. hat darauf hingewiesen, wenn man die Energieniveaus im Benzol oder einem ähnlichen System ausrechnet, daß dann energetisch gesehen, keine Trennung zwischen s - und p -Niveaus besteht, daß also die s -Niveaus energetisch teilweise ähnlich wie die p -Niveaus liegen. Aber das hat mit der s - / p -Trennung im eigentlichen Sinne wenig zu tun. Und weil eben früher reine p -Elektronen Theorien so wichtig waren, wie z.B. diejenige von Pariser-Parr-Pople, hatten wir uns mit dieser Frage beschäftigt. Unsere Arbeit wurde wohl eher von Theoretikern als Organikern zur Kenntnis genommen.
Es gab einen entscheidenden Durchbruch, ich glaube das steht auch in meinem Buch (16), in der Anerkennung der Theorie durch die experimentelle Organischen Chemie, das waren die Woodward-Hoffmann Regeln. Mit den Woodward-Hoffmann Regeln haben die organischen Chemiker zum ersten mal die Wichtigkeit der Theorie eingesehen. Von da ab war es auch einfacher, mit den Kollegen aus der Organischen Chemie zu reden. Ich meine, heute ist es überhaupt kein Problem mehr, denn die heute verfügbaren Rechnungen überzeugen einfach. Früher war ja das Problem: man konnte nur Hoffnungen machen, man konnte sagen: also wenn wir tüchtig weiter arbeiten, dann werden wir es vielleicht mal eines schönen Tages schaffen - Ergebnisse zu liefern, mit denen ihr auch etwas anfangen könnt. Und dies ist inzwischen eingetreten.
A: Glauben Sie, daß man eines Tages sogar Schmelz- und Siedepunkte berechnen könnte?
K: Das ist nicht so sehr eine Frage der Quantenchemie als der Thermodynamik. Sie müssen Freie Energien von Flüssigkeiten und Festkörpern und Gasen ausrechnen. Und das ist im Prinzip möglich. Bei idealen Gasen steht es in jedem Lehrbuch, wie man das macht. Und wir haben uns auch seinerzeit mal mit thermodynamischen Fragen beschäftigt (17). Man kann das sehr wohl machen. Nur ist es eben nicht mehr reine Quantenchemie. Das gleiche gilt für Übergangstemperaturen, wenn Sie also mehrere Phasen haben - - . Bei welcher Temperatur beispielsweise die Freie Energie der beiden Phasen gleich ist - da haben Sie den Übergangspunkt. Und eine Sache, an der z.B. die Festkörperphysik sehr interessiert ist, das sind die Übergangstemperaturen von Supraleitern. Daran wird intensiv gearbeitet. Man weiß im Grunde, was man dazu verstehen muß, da muß man die Phononenspektren genau kennen.
Also da sehe ich keine grundsätzliche Schwierigkeit, es kommt darauf an, daß wir die erforderlichen Größen mit hinreichender Genauigkeit bekommen. In der Flüssigkeit sind auch die zwischenmolekularen Wechselwirkungen wichtig, anders als in der Gasphase.
A: Gibt es unter diesen Aspekten für die semiempirische Methoden noch Hoffnungen?
K: Das ist schwer zu beantworten. Aber ich kann Ihnen z.B. sagen, das HF ist ein thermodynamisch sehr interessantes System, weil es auch in der Gasphase noch sehr stark assoziiert ist. Hierzu hat Herr Suhm in Zürich, in der Gruppe von Herrn Quack kombinierte experimentelle und theoretische Untersuchungen gemacht, ausgehend wirklich von den intermolekularen Wechselwirkungen zwischen HF-Molekülen, um die thermodynamischen Eigenschaften vom flüssigen zum gasförmigen HF zu bestimmen. Das sind Dinge, die im Prinzip gehen. Bei der Anwendung von semiempirischen Methoden bin ich ein bißchen skeptischer, aber man kann sich da natürlich durch das Drehen an Parametern immer behelfen - -
Dewar, der große Mann der semiempirischen Theorie, hat die Größen, die er ausgerechnet hat, korreliert mit Enthalpien bei Zimmertemperatur. Normalerweise, wenn man quantenmechanisch rechnet, betrachtet man Energien am absoluten Nullpunkt. Bei ihm waren alle thermodynamischen Effekte in seiner Parameterisierung mit drin.
A: Wie finden Sie Dewars Buch A Semiempirical Life (18)?
K : Ich hab das nie gelesen. Ist es vor kurzem erschienen?
A: 1992.
K: Ich kann dazu nichts sagen. Ich habe Dewar persönlich gekannt. Er war ein sehr faszinierender Mensch, muß ich sagen, sehr an Wissenschaft interessiert - -
A: Es ist interessant das von Ihnen zu hören und Sie haben ja auch in Ihrem Buch der Hückel Didaktik viel Raum eingeräumt.
K: Vielleicht kann ich noch sagen, als ich mein Buch geschrieben habe (3a), da waren die semiempirischen Theorien noch sehr im Schwang. Und ich habe mich bemüht, eine Begründung und eine Rechtfertigung für semiempirische Methoden zu formulieren. In meinem Buch gibt es auch ein relativ langes, schwer verständliches Kapitel, zum Teil basierend auf der Doktorarbeit von Herrn Driessler, wo wir versucht haben, aus ab initio Rechnungen semiempirische Parameter herzuleiten (19). Etwas ähnliches hat auch Karl Fried in Chicago gemacht. Der kann heute wirklich effektive Hamilton-Operatoren ausrechnen, wo er ab initio-berechnete semiempirischen Parameter mit solchen identifizieren kann, die in der semiempirischen Theorie eine Rolle spielen. Vielleicht hat dieser Anschluß der semiempirischen an die ab-initio-Theorie durchaus Zukunft.
Aber der Zug ist eigentlich jetzt in eine andere Richtung gefahren. Heute sind sehr erfolgreich sogenannte Dichtefunktional-Methoden (24-26). Und die könnten semiempirische Methoden fast überflüssig machen, weil sie im Rechenaufwand soviel sparsamer sind, als die herkömmlichen ab initio Methoden und trotzdem recht gute Ergebnisse liefern. Das ist sozusagen ein Trend in neuerer Zeit.
Sie haben meinen Artikel über Hund in der Angewandten Chemie gelesen (1)?
A: Leider noch nicht.
K: Er hatte den Titel: Friedrich Hund und die Chemie. Ferner hatte ich einen Vortrag gehalten zum 100. Geburtstag von Hückel. Das war in Marburg vor einem dreiviertel Jahr. Ich habe mir überlegt, ob ich den vielleicht zu einen Artikel für die Angewandte Chemie umwandle - aber das kostet Zeit. Der Vortrag hatte den Titel Was mir an der Hückel Näherung gefällt. Alles was in der Chemie mit Delokalisation zu tun hat, ist ja mit der Hückel Theorie ganz gut zu beschreiben. Aber dabei wird ja auch deutlich, daß sie nicht der Weisheit letzter Schluß sein kann.
Ich überlege noch, wer die heutigen Protagonisten der semiempirische Theorie sind. Es gibt ja Kollegen, die sie noch betreiben. Dazu gehört z.B. Michael Zerner in Gainsville, Florida. Der hat eine Methode erfunden, genannt ZINDO (man muß also unterscheiden, es gibt auch eine von Jug, SINDO). Jug muß man hier natürlich auch nennen. ZINDO ist eine Methode, die auch für Übergangsmetallkomplexe von organischen Systemen geeignet ist. Ein anderer hier zu nennender Kollege ist Walter Thiel in Zürich, der auch noch erfolgreich Semiempirie betreibt, der z.B. ganz große Fullerene wie
![]() und so berechnet hat. Er war ein früherer Mitarbeiter von Dewar und ist verantwortlich für einige Verbesserungen, die damals in der Gruppe gemacht wurden, z.B. die MNDO-Methode. Er beherrscht aber auch die ab initio Verfahren. Freed wurde schon genannt, er ist jemand, der sich mit vielen Dingen beschäftigt, unter anderem auch mit der Rechtfertigung und der physikalischen Bedeutung der semiempirischen Parametrisierungen. Leider hat man, wenn man versucht, auf dem Prinzip der effektiven Hamilton Operatoren eine semiempirische Theorie herzuleiten, das Problem daß dann Wechselwirkungen höherer Ordnung auftreten, zum Beispiel 3-Teilchen Wechselwirkungen, die man dann irgendwie wegsimulieren muß.
und so berechnet hat. Er war ein früherer Mitarbeiter von Dewar und ist verantwortlich für einige Verbesserungen, die damals in der Gruppe gemacht wurden, z.B. die MNDO-Methode. Er beherrscht aber auch die ab initio Verfahren. Freed wurde schon genannt, er ist jemand, der sich mit vielen Dingen beschäftigt, unter anderem auch mit der Rechtfertigung und der physikalischen Bedeutung der semiempirischen Parametrisierungen. Leider hat man, wenn man versucht, auf dem Prinzip der effektiven Hamilton Operatoren eine semiempirische Theorie herzuleiten, das Problem daß dann Wechselwirkungen höherer Ordnung auftreten, zum Beispiel 3-Teilchen Wechselwirkungen, die man dann irgendwie wegsimulieren muß.
Wie schon gesagt, gibt es auch die sogenannten rein empirischen Methoden, wie Kraftfeldmethoden und Molecular Modelling. Diese haben natürlich eine sehr große Bedeutung. Es gibt verschiedene Kraftfeldprogramme. Eines davon ist das GROMOS Programm, das ist also im wesentlichen von van Gunsteren an der ETH Zürich. Dann gibt es CHARM, 'chemistry at Harvard', das ist von Martin Karplus. Ein weiteres Programm heißt AMBER. Es gibt noch weitere Autoren, die auf dem Kraftfeldsektor eine wichtige Rolle spielen, zum Beispiel Peter Kollman in San Francisco und natürlich Allinger (27) in Athens, Georgia, ferner Lifson in Israel. Kraftfeld-Ansätze werden auch in Ihrem HyperChem, und vor allem für Proteine in Molecular Modelling Simulationen verwendet. In der chemischen Industrie spielt das eine sehr große Rolle. Ein ehemaliger Mitarbeiter von mir, Michael Schindler, der bei uns die IGLO Methode entwickelt hat, macht jetzt Molecular Modelling bei BAYER.
Neuerdings verwendet man auch Kombinationen verschiedener Methoden (Stichwort embedding), zum Beispiel von Morukuma in Atlanta. Denken Sie sich irgendein größeres System, eine ganze Menge von Molekülen, Lösungsmittel und so weiter und teilen Sie das sozusagen in viele Kästen auf. Sie machen im inneren Kasten eine ab initio Rechnung, dann im etwas größeren Bereich rechnen Sie semiempirisch, noch weiter draußen mit Molecular Modelling. Derartige Einbettungsverfahren haben vermutlich eine Zukunft.
A: Ich erwähnte vorhin die Autobiographie von Dewar. Wie finden Sie die von Hückel (20)?
K: Sie ist ein eher schwaches Buch. Sie hätte von einem Lektor redigiert werden müssen. Darin steht zu viel Unbedeutendes, Irrelevantes. Es gibt eine Autobiographie von Mulliken (21) - -
Man findet auch immer wieder kuriose Dinge in der Geschichte der semiempirischen Theorie, z.B. die sogenannte Methode von Hückel und Hartmann (22 a-c), ich weiß nicht, ob Sie davon gehört haben. Sie ist auch zu Recht vergessen. Aber damit hat Hartmann damals, in seinen späten Jahren, viel Wind gemacht.
A: Dewar trat ja auch mal daneben mit seiner split orbital Methode. Er beschreibt das anschaulich in seiner Autobiographie (23).
K: Sieht er jetzt ein, daß er Unrecht gehabt hat? So ging es tatsächlich nicht. Er wollte ja die Reduktion der Elektronenwechselwirkung im Modell von Pariser, Parr, Pople verständlich machen, und da hat er sich gedacht, er halbiert die p -Orbitale in obere und untere Lappen - das wäre alles wunderschön gewesen, wenn er nicht das Orthogonalitätsproblem völlig übersehen hätte.
A: Er gibt es zu und schiebt den Fehler auf seine argentinische Mitarbeiterin - -
K: Dewar war, ich hab es schon erwähnt, ein sehr faszinierender Mann. Über Wissenschaft konnte man mit ihm stundenlang reden. Er war ein echter Engländer. In Amerika, da wahren die Engländer ihre nationalen Eigenarten mehr als andere Ausländer. Eines seiner Probleme war folgendes. Er war lange Zeit ein großer Bewunderer von Pople, seine Arbeiten bauen ja auf denen von Pople auf, das ist unbestreitbar. Aber er hat es Pople sozusagen nie verziehen, daß er das Feld verlassen hat und von der semiempirischen Theorie in die ab initio Theorie übergewechselt ist.
A: Pople hat dann auch später wenig Gutes über die semiempirische Theorie gesagt.
K: Aber Pople hat vorher wesentlich zu den semiempirische Methoden beigetragen!
A: Das möchte er heute wohl nicht mehr so sehr hören?
K: Er hört es möglicherweise nicht mehr so gerne. Und wenn ich da in meinem Buch von der Pople Näherung rede, wird er dem vermutlich nicht zustimmen. Er hat aber vermutlich mein Buch nicht gelesen, es ist ja in deutsch abgefaßt. Ich würde heute auch nicht mehr von der Pople Näherung sprechen. Aber damals, als ich das geschrieben habe, war der Name Pople mit der CNDO Geschichte eng verbunden (28).
A: Longuet-Higgins verließ aprupt die Quantenchemie, wo mag er sein?
K: Das kann ich Ihnen sagen: an der Universität von Sussex, - in der Neuroinformatik oder so ähnlich. Longuet-Higgins war sicher ein sehr bedeutender und interessanter Mann, der mit Coulson zusammen seinerzeit wichtige Arbeiten zur Hückel Theorie gemacht hat, seinerzeit. Er hat noch weitere Verdienste, die mit der Hückel Theorie nicht so viel zu tun haben. Er hat z.B. die Symmetriegruppen nicht-starrer Moleküle eingeführt. Diese ist wichtig für die Theorie der Rotations-Schwingungsspektren. Und dann hat er noch eine grundlegende Arbeit zusammen mit Herzberg über die sogenannte geometrische Phase veröffentlicht, über das, was man heute als Berry-Phase bezeichnet. Daß bei einer vollen Drehung um einen singulären Punkt das Vorzeichen der Wellenfunktion sich ändert, ist eine ganz ulkige Geschichte. Insofern ist Longuet-Higgins schon ein wichtiger Mann, aber er hat das Gebiet der Theoretischen Chemie leider verlassen.
Was man ihm übrigens zum Vorwurf machen muß ist, daß er die Numerik völlig abgelehnt hat und daß er S.F. Boys, der ein kleiner Dozent an seinem Lehrstuhl war, das Leben außerordentlich schwer gemacht hat. Boys ist ja einer der wichtigsten Pioniere der ab initio Quantenchemie.
A: Herr Professor Kutzelnigg, ich bedanke mich für dieses Interview.
Referenzen und Anmerkungen
(1) W. Kutzelnigg, Friedrich Hund und die Chemie. Angew. Chem. 1996, 108, 629-643.
(2) z.B. W. Kutzelnigg und R. Mecke, Die Struktur und das Schwingungsspektrum des
Harnstoff-Kations in normalen und anormalen Salzen.
Chem. Ber. 1961, 94, 1706-1718 und Ref. dort.
(3a) W. Kutzelnigg, Einführung in die Theoretische Chemie. 2 Bände.
Verlag Chemie, Weinheim, 1975 bzw. 1978.
(3b) W. Kutzelnigg, Einführung in die Theoretische Chemie. 2 Bände.2. Auflage.
Verlag Chemie, Weinheim, 1992 bzw. 1994.
(4) W. Kutzelnigg, Zur Verwendung der vollständigen Laguerre-Funktionen bei quanten-
chemischen Rechnungen. Theoret. Chim. Acta, 1963, 1, 257-267.
(5) W. Kutzelnigg, Die Lösung des quantenmechanischen Zwei-Elektronenproblems durch
unmittelbare Bestimmung der natürlichen Einelektronenfunktionen. I.
Theorie. Theoret. Chim. Acta, 1963, 1, 327-342.
(6) W. Kutzelnigg, Résolution du problème à deux électrons en mécanique quantique par
détermination directe des orbitales naturelles. II: Application aux états
fondamentaux de l'hélium et des ions isoélectroniques.
Theoret. Chim. Acta, 1963, 1, 343-352.
(7) IBM Deutschland, Sindelfingen gibt dazu folgende Daten:
IBM 1620: transistor technology; 20 KB storage of 6 bit each; 1 electric control panel
for automatic or manual work; 1 typewriter for input and output, 10 Byte/sec.
Programmable in:
a) SPS (Symbolic Programming System),
b) FORTRAN - one of the earliest versions; subroutines were available.
IBM 1623: memory unit for up to 60 KB total.
IBM 1621: papertape reader, 8 channels, 150 Byte/sec.
IBM 1624: papertape punch, 15 Byte/sec.
IBM 1622: card reader, 80 coulumns, max. 250 Byte/sec.
card punch, 125 cards/min.
(8) W. Kutzelnigg, Zur Behandlung der Bindungsalternierung als Störung der Hückel'schen
MO-Theorie. Theoret. Chim. Acta, 1966, 4, 417-433.
(9) Interview mit R. Daudel am 3.6.1997.
(10) C.A. Coulson, Hückel Theory for Organic Chemists. Academic Press, London, 1978. p. 171: "Someone said that if you ask me a question before dinner I am an optimeist - after dinner, I am a pessimist."
(11) HyperChem for Windows. Hypercube Inc., Canada, 1994.
(12) L. Pauling and E.B. Wilson, Introduction to Quantum Mechanics.
McGraw-Hill, New York, 1935
(13) Ref 3a-b, Bd.II, p. 4, und Interview mit R. McWeeny.
(14) Ref 3a-b, Bd.II, p. 1.
(15) W. Kutzelnigg, G. Del Re and G. Berthier, s - and p -Electrons in Theoretical Organic Chemistry. in: s - and p -Electrons in Organic Compounds. Fortschritte der chemischen Forschung,(Topics in Current Chemistry), Vol. 22. Springer Verlag, Berlin, 1971.
(16) Ref 3a-b, Bd.II, p. 141.
(17) W. Kutzelnigg and R. Ahlrichs, Theoret. Chim. Acta, 1968, 10, 377.
(18) M.J.S. Dewar, A Semiempirical Life. Amer. Chem. Soc., Washington, DC, 1992.
(19) Ref. 3a-b, Bd.II, p. 91 und F. Driessler und W. Kutzelnigg,
Theoret. Chim. Acta 1977, 43, 307.
(20) E. Hückel, Ein Gelehrtenleben. Verlag Chemie, Weinheim, 1975.
(21) R. Mulliken, Life of a Scientist. Springer Verlag, Berlin, 1989.
(22a) H. Hartmann, Zur Theorie der p -Elektronensysteme. Z. Naturforsch. 1960,15a, 993-1003.
(22b) E. Ruch, Zur Theorie der p -Elektronensysteme. Z. Naturforsch. 1961,16a, 808-815.
(22c) siehe dazu: G. Hohlneicher und G. Scheibe, Die erweiterte Hückelsche p -Elektronentheorie und ihre Anwendung auf einfache Konjugationssysteme. Tetrahedron, 1963, Suppl. 2, 189-200.
(23) Ref 18, p. 106
(24) R.G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules.
Oxford University Press, New York, 1989.
(25) E.S. Kryachko and E.V. Ludeña, Energy Density Functional Theory of Many Electron
Systems. Kluwer Acad. Publ., Dordrecht, 1990.
(26) J.K. Labanowski and J.W. Andzelm (Eds.), Density Functional Methods in Chemistry.
Springer, New York, 1991.
(27) N.L Allinger, Molecular Mechanics. Amer. Chem. Soc., Washington, DC, 1982.
(28) J.A. Pople and D.L. Beveridge, Approximate Molecular Orbital Theory.
McGraw-Hill, New York, 1970.
(29) Ref 3a-b, Bd.II, Kapitel 11.11, p. 308.